16 - cSCC - Hide and Seek
The most dangerous skin cancer has a secret: when treatment comes looking, it pulls its survival switch inside the cell — beyond the reach of every approved drug. Until now.


Your point of view is vast — inside something alive that should not exist, hiding in plain sight, dangerous. The surroundings are dark machinery stretching away into depth. At the periphery, warm amber light bleeds through a threshold that has no hard edge — a horizon rather than a wall. On the other side, large Y-shaped forms press against it, trying to pass through, prevented from reaching the danger within. They remain there, inactive, softly luminous, slightly blurred. Deep in the cold interior, a single point of amber light burns quietly in the dark. Small. Faint. Something repurposed. It does not announce itself. It simply burns there — the heartbeat of an intruding cell.
Every drug approved for this disease is on the other side of that horizon. None of them can pass through. But that light within must be stopped.
It is known as C5aR1. It normally lives at the surface of the cell, where it can be reached. Starved of oxygen, it retreats inside — suppressing the death programs that should end this cell’s existence, represented here in the form of light, keeping alive something that should already be gone. The cell divides. Then divides again. Four cells become eight. Eight become a tumor. The tumor grows until, in time, it kills the person it lives in.
That sequence begins with a signal in the dark. This article is about stopping it.
Image created using ChatGPT (OpenAI) from a prompt developed with Claude (Anthropic).
Last updated: April 19, 2026
Researched and written in collaboration with Claude (Anthropic), enhanced by a structured knowledge base built from years of the author’s primary research into complement biology and the C5a signaling pathway. Claims are labeled for accuracy throughout; the legend below explains the system.
Legend
✓ Established science — peer-reviewed, reproducible, not contested in literature
~ Supported interpretation — grounded in real data; involves analytical judgment
? Speculative — directionally reasoned; forward-looking or unconfirmed
1: The Disease
1.1 Living with cSCC
Cutaneous squamous cell carcinoma (cSCC) is the second most common skin cancer worldwide and the most common skin cancer to spread to other parts of the body, with approximately one million new cases diagnosed in the United States each year. ✓ That number is rising — European registry data projects an annual increase in cSCC incidence of 2.4% to 5.7% per year, driven by an aging population, cumulative lifetime UV damage now manifesting in older cohorts, an expanding population of immunosuppressed patients on long-term medications, and improved detection catching cases that previously went undiagnosed. ~ The global market for cSCC treatment was valued at approximately $13.7 billion in 2024 and is projected to reach $27.5 billion by 2034. ~ This is not a static disease burden. It is a growing one.
Most of those cases are caught early. The lesion is excised. The patient goes home. For the vast majority, that is the end of the story.
For approximately 5% of cases — those whose disease has progressed beyond the reach of surgery — the story is just beginning. ~
One such story unfolded in September 2022, when a 79-year-old woman enrolled in a clinical trial at the University Hospital Erlangen in Germany. Her cSCC had first appeared on her scalp. It was excised surgically in 2019. In November 2021, it came back. She received 144 days of treatment with cemiplimab — a drug that works by releasing a brake on the immune system’s cancer-fighting cells (called the T-cell checkpoint) that tumors learn to engage. It was approved specifically for this disease and represents one of the best tools medicine currently has for advanced cSCC. The result: progressive disease. The tumor continued to grow. ✓
Subsequently, she was enrolled into a trial of vilobelimab — an antibody that works by neutralizing C5a, the protein that binds to a receptor called C5aR1 and drives the inflammatory signals that were fueling her tumor’s growth and suppressing her immune response. Vilobelimab blocks C5a before it can reach the receptor. INF904 (izicopan), the drug at the heart of this article, blocks the receptor itself. Same upstream signal. Same story. A better ending — as we will see. She received infusions approximately every two weeks for nearly two years — 46 infusions in total.
By Cycle 6, her tumor was gone. A biopsy confirmed it. The complete remission lasted through Cycle 12. Her overall response — periods of confirmed partial or complete remission — lasted 512 days, with at least 418 of those days as confirmed response. No drug-related adverse effects were reported across the entire treatment course. ✓
She was 79 years old. She had already failed the best drug the field had to offer. And she achieved a biopsy-confirmed complete remission on an anti-inflammatory antibody.
That case is the beginning of this article. It is also, depending on what happens next, the beginning of something larger.
1.2 What cSCC Is
cSCC is the malignant transformation of keratinocytes — the cells that make up the outer layer of the skin. It is the most dangerous of skin cancers, whereas basal cell carcinoma, which is far more common, almost never spreads to other parts of the body and almost never kills. cSCC both spreads and kills.
Risk factors include cumulative sun damage, immunosuppression, chronic skin inflammation, and prior radiation treatment to the same area of skin — the kind of chronic inflammatory damage that the drug at the center of this article is designed to interrupt. Patients who have received solid organ transplants and are on immunosuppressive drugs have dramatically elevated cSCC risk and far worse outcomes when it develops. This is the first clinical signal that the immune system’s state is not just a background condition in cSCC. It is a driver. ✓
cSCC incidence is rising at 2.4% to 5.7% annually. ~ The disease disproportionately affects elderly patients — most lesions appear on chronically sun-damaged skin of the scalp, face, ears, and hands in patients in their seventh and eighth decades of life. Many early lesions go unexamined; the one million annual US diagnoses represent confirmed cases, and the true incidence is likely higher. ~
Most cSCC cases are caught early. Simple excision — a brief outpatient procedure under local anesthetic costing a few hundred to a few thousand dollars — is curative in the majority of low-risk cases. Mohs surgery, a more specialized technique used for higher-risk locations, achieves similarly high cure rates. Surgery is the right tool for early disease and it works well. The unmet need this article addresses begins precisely where surgery is no longer an option — when the lesion has grown too large, invaded too deeply, or spread to lymph nodes or distant sites. That transition from manageable to life-threatening affects approximately 15,000 to 20,000 Americans per year whose disease becomes locally advanced, and 7,000 to 10,000 more whose disease metastasizes. For the subset with high-risk features — large tumors, perineural invasion, immunosuppressed patients — recurrence rates after surgery can reach 20 to 30%, meaning some patients initially treated successfully will eventually enter the advanced disease population. ~
In and around cSCC tumors, the immune system’s first-responder white blood cells — neutrophils, which normally seek out and destroy threats like bacteria and damaged cells — are present in high numbers. But they are not doing their job. They are, as we will see, doing something far more dangerous.
1.3 The cSCC Story at a Glance
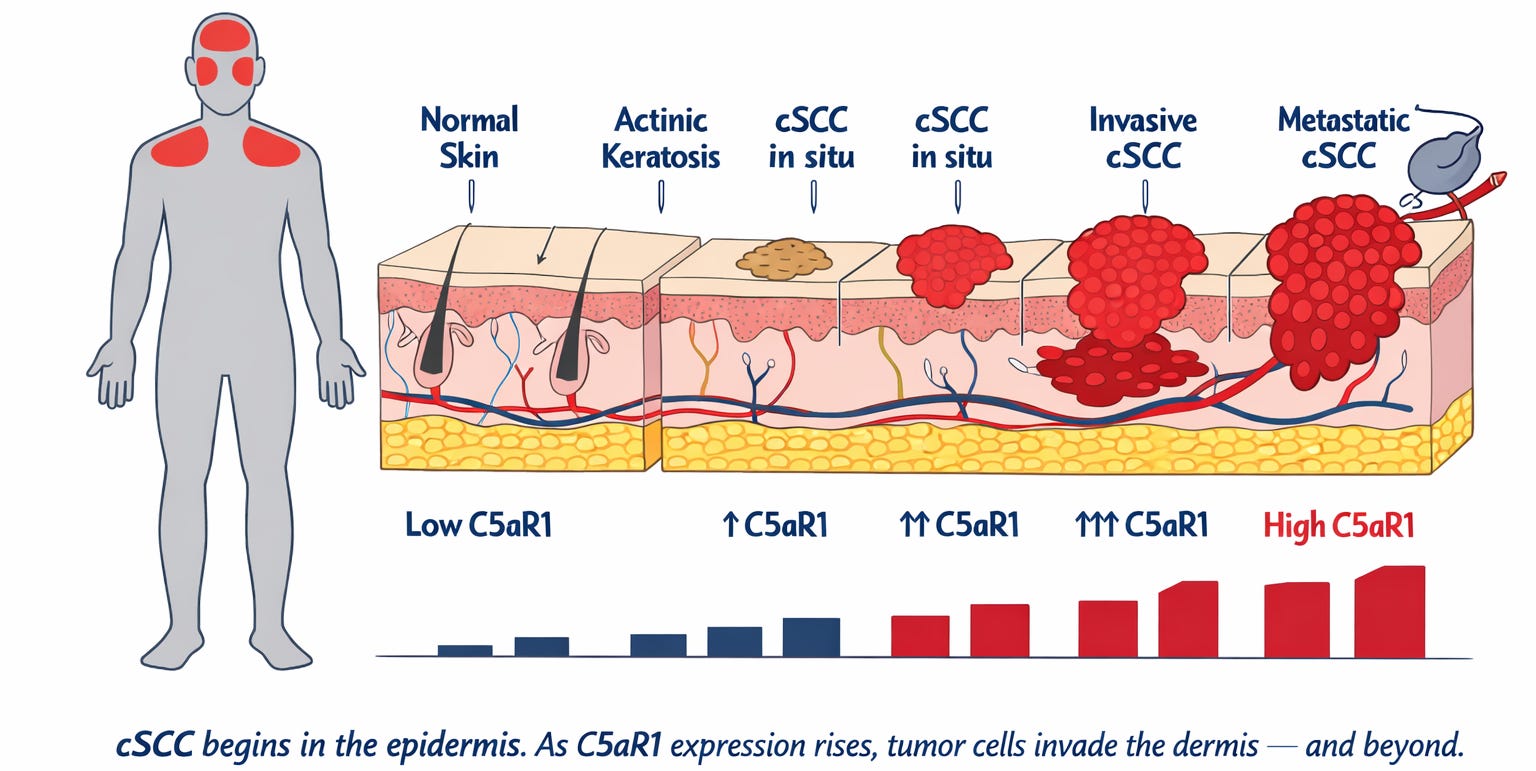
Figure 1a. The human figure on the left marks where cSCC develops — on sun-exposed skin, most commonly the face and scalp. The cross-section shows the skin’s layers from the outermost surface (the epidermis — the thin protective outer layer where cSCC originates) down through the dermis (the deeper tissue layer the tumor invades as it advances) to the subcutaneous tissue beneath. Beside each disease stage, a bar reflects the level of a receptor — C5aR1 — found on tumor cells at that stage. Normal skin carries almost none. At every step toward metastatic cancer, the level rises. The higher the stage, the worse the prognosis. That receptor is what this article is about.
cSCC in situ (surface-confined) is shown at two severity levels reflecting the clinical spectrum before the cancer begins growing deeper into the skin.
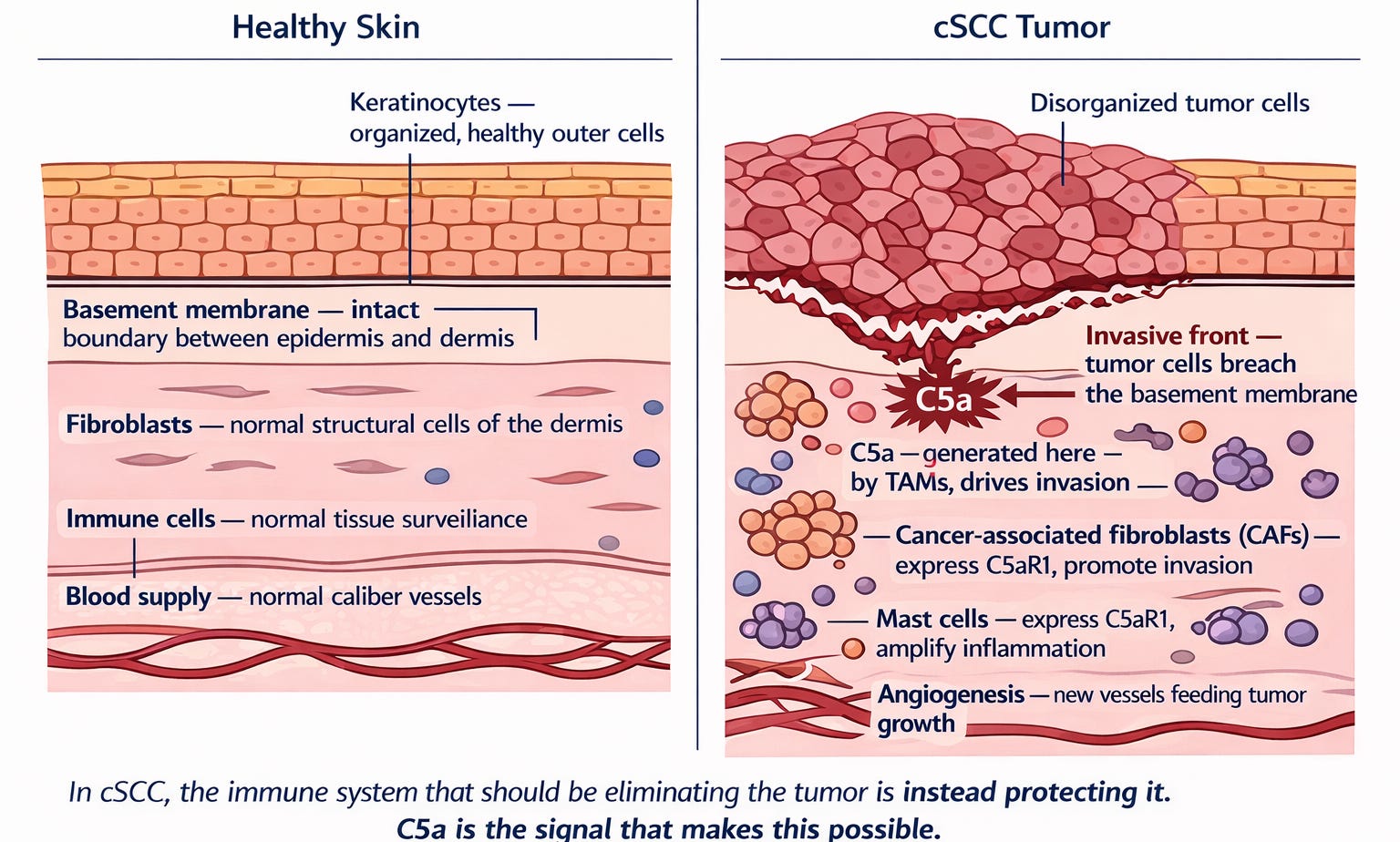
Figure 1b. Two cross-sections of skin at the same scale. Left panel: healthy skin. Keratinocytes arranged in organized layers, the basement membrane intact, immune cells sparse, the dermis quiet. Right panel: cSCC. The organized architecture is gone. The basement membrane has been breached at the invasive front. Macrophages expressing uPA are generating an inflammatory signal that drives everything that follows. Fibroblasts have been reprogrammed into agents of invasion. New blood vessels have formed to feed the growing tumor. The inflammatory signal driving every one of these changes has a name — C5a.
In cSCC, the tumor learns to generate its own inflammatory signal using the immune cells already present in the skin. Those immune cells — macrophages — produce an enzyme called urokinase (uPA) that cuts a complement protein called C5 into a fragment called C5a. C5a then binds to a receptor — C5aR1 — on macrophages, other immune cells, and the tumor cells themselves. The result is a tumor microenvironment in which the immune system that should be killing the tumor is instead protecting it. ✓
But there is a second mechanism. One that was only described in peer-reviewed literature in July 2025. One that explains the title of this article — and why the drug that could change the story for the patients who need it most has never yet been given to a patient with cSCC.
To understand both mechanisms — the one medicine has been trying to address and the one it has been missing entirely — we need to follow C5a into the biology. Section 2 explains precisely how the signal is generated, what it does to every cell it touches, and why the treatments that currently exist have never once addressed it where it ultimately hides.
2: The Biology
2.1 The Components
The cSCC story takes place in a defined and bounded space. Below is the complete cast of structures, cells, and molecules that appear in this article.
The barrier structures:
Epidermis — The outer skin layer, composed of keratinocytes — the malignant keratinocyte (tumor cell) is the central character of this story. The physical boundary that C5a-driven tumor cells breach as they invade deeper into the body.
Basement membrane — The thin structural boundary separating the epidermis from the dermis beneath it. Intact in healthy skin. Its breach at the invasive front is the defining event of invasive cSCC.
Dermis — The connective tissue layer beneath the epidermis. Contains fibroblasts, blood vessels, and immune cells — the invasion target for advancing cSCC cells.
Extracellular matrix (collagen I) — The structural protein scaffold of the dermis. C5a-stimulated cSCC cells actively invade through this scaffold — confirmed in Heiskanen’s 3D spheroid model.
Hypoxic tumor core — The low-oxygen center of solid tumors. As a cSCC tumor grows faster than its blood supply, the interior becomes severely oxygen-deprived. This is the most therapy-resistant and prognostically dangerous compartment of the tumor — and the site of the intracellular C5aR1 survival program.
Plasma membrane — The outer boundary of individual cells, including malignant keratinocytes (tumor cells). A thin, selectively permeable layer that controls what enters and exits the cell. Antibodies and peptides cannot cross it. Cell-permeable small molecules can. The plasma membrane is the threshold between the drug-saturated world outside the cell and the intracellular C5aR1 survival program within.
The cellular players:
Keratinocytes — The dominant cell of the epidermis and the cell of origin for cSCC. When they undergo malignant transformation, they become the tumor cells. They express C5aR1 when in contact with cancer-associated fibroblasts.
Cancer-associated fibroblasts (CAFs) — Connective tissue fibroblasts surrounding the tumor, reprogrammed by tumor signals. In cSCC they express C5aR1. Their proximity to tumor cells induces C5aR1 expression in the cancer cells themselves. C5aR1-positive CAFs independently predict poor 5-year survival.
Tumor-associated macrophages (TAMs) — The primary myeloid driver of C5a signaling in the tumor. Myeloid cells are the family of immune cells that includes macrophages, neutrophils, and other first-responder white blood cells. TAMs express urokinase (uPA), an enzyme that cleaves complement C5 to generate C5a via the fibrinolytic system — bypassing classical complement pathways entirely. C5aR1-positive TAMs suppress cancer-killing T cells, promote new blood vessel formation, and drive tumor progression.
Tumor-associated neutrophils (TANs) — Neutrophils recruited into the tumor microenvironment and reprogrammed by C5a-driven signals into a pro-tumor phenotype. Unlike their role in hidradenitis suppurativa, pyoderma gangrenosum, or ANCA-associated vasculitis — where neutrophils are the primary effectors causing direct tissue destruction — in cSCC they are corrupted into TANs that suppress CD8+ T cells, promote angiogenesis (new blood vessel formation), and release NET-associated proteases that degrade the basement membrane and facilitate invasion. Same cell. Corrupted function.
CD8+ T cells — The immune system’s cancer-killing lymphocytes. Rendered exhausted and dysfunctional by C5aR1-driven myeloid suppression. When C5aR1 is blocked, they recover their cancer-killing function.
Myeloid-derived suppressor cells (MDSCs) — Immunosuppressive myeloid cells recruited by C5a. They reinforce the immunosuppressive tumor microenvironment alongside TAMs, restraining T-cell anti-tumor activity.
Mast cells — Tissue-resident innate immune cells that express C5aR1 in cSCC. They amplify the inflammatory signal TAMs initiate — releasing mediators that reinforce immunosuppression and drive angiogenesis alongside TAMs.
The molecular players:
Complement C5 — The complement protein cleaved by uPA-expressing macrophages to generate C5a. C5 is also stored in intracellular vesicles within tumor cells, where it can be cleaved internally by cathepsin D to generate C5a inside the cell — creating an autocrine intracellular loop. This mechanism is confirmed in cancer cells broadly; its application to cSCC specifically is a supported inference. ~
C5a — The central villain. Generated in the cSCC tumor microenvironment by uPA+ macrophages cleaving complement C5 via the fibrinolytic system — bypassing classical complement. Also generated inside tumor cells via cathepsin D cleavage of intracellular C5 stores. ~ Binds C5aR1 to drive every downstream pro-tumor program.
C5aR1 — The receptor and the target. Expressed on TAMs, mast cells, CAFs, neutrophils, and malignant keratinocytes. Expression tracks stepwise through every stage of cSCC progression and independently predicts metastatic risk and poor 5-year disease-specific survival. Under hypoxia (severe oxygen deprivation), C5aR1 is drawn inside tumor cells where it drives survival from within — independently of extracellular C5a.
Urokinase (uPA) — An enzyme expressed by macrophages in the tumor that cleaves complement C5 to generate C5a via the fibrinolytic system — bypassing upstream complement proteins entirely. This makes anti-C5 antibodies pharmacologically irrelevant to C5a generated in the cSCC tumor microenvironment.
PD-1 / PD-L1 — The checkpoint proteins targeted by all three approved drugs in advanced cSCC. PD-L1 is the key — displayed on the surface of cancer cells. PD-1 is the lock — sitting on the surface of T cells. When PD-L1 finds PD-1, the T cell receives a stand-down signal. Checkpoint inhibitors block either the key (anti-PD-L1) or the lock (anti-PD-1), enabling T cells to remain in an active state — free to recognize and attack the tumor. The reader already knows this mechanism from C5a and C5aR1: same lock-and-key logic, different players, different step in the cascade.
VEGF — The signal that drives new blood vessel formation feeding tumor growth. Produced downstream of C5aR1-activated macrophages and mast cells. Blocking C5aR1 blocks VEGF upregulation — upstream of anti-VEGF drugs that address only this one downstream consequence.
NF-κB — The pro-survival molecular switch inside cells. In malignant keratinocytes, C5aR1 signaling keeps NF-κB in a pro-survival configuration. C5aR1 blockade shifts it toward programmed cell death specifically in tumor cells, not normal tissue.
NETs (neutrophil extracellular traps) — Web-like structures of DNA and protein released by activated neutrophils and TANs. In cSCC, NET-associated proteases degrade the basement membrane and extracellular matrix, creating physical corridors for tumor cell invasion. NETs also release DAMPs — cellular alarm signals — that activate complement, generating more C5a and feeding the self-amplifying cycle.
INF904 — The hero of this story. InflaRx’s oral, selective small molecule C5aR1 inhibitor. Cell-permeable by its drug class — meaning it crosses the plasma membrane and reaches intracellular C5aR1 that antibodies cannot access. No Boxed Warning. Addresses both surface C5aR1 and the intracellular survival program in the hypoxic tumor core.
2.2 The Smoking Gun: Deadlier the Tumor, Higher the C5aR1
Before explaining how the mechanism works, it is worth pausing on what the field’s own data has already shown. The thesis of this article does not begin with speculation. It begins with published human pathology — and with a receptor that keeps appearing, at every stage of disease, in every patient most likely to die.
In 2025, a research team in Finland published the largest systematic study of C5aR1 in human cSCC tissue ever conducted. ✓ Led by Lauri Heiskanen and colleagues at the University of Turku, the study examined 174 invasive cSCCs, 62 normal skin samples, 53 samples of a precancerous condition called actinic keratosis (AK), 47 samples of cSCC confined to the surface layer, 9 metastatic tumors, and a large group of primary tumors whose outcomes over time were known. ✓
The findings were unambiguous. ✓
C5aR1 expression increased at every step of disease progression: normal skin to AK to surface-confined cSCC to invasive cSCC to metastatic cSCC to the most aggressive form of the disease. The receptor did not merely mark disease. It marked severity. ✓
C5aR1 expression on the surface of tumor cells independently predicted poor disease-specific 5-year survival. The statistical separation was significant at p < 0.0001, with visible separation beginning at the three-year mark. ✓
C5aR1 expression in the cancer-associated fibroblasts surrounding the tumor also independently predicted poor 5-year survival. p < 0.0001. Both markers are independent prognostic predictors. ✓
And in a three-dimensional cell culture model of tumor invasion, recombinant C5a directly drove cSCC cell invasion through collagen. The statistical significance was p < 0.0001 at 96 hours. ✓
The authors also found that cSCC cells do not express C5aR1 when grown in isolation. The receptor only appears when tumor cells are in contact with fibroblasts. C5aR1 emerges at the invasive edge, where tumor cells make contact with their environment. ✓
The conclusion the field’s own pathology draws is direct: C5a signaling — and specifically C5aR1, the receptor that receives and transmits it — is the upstream driver of the biological behaviors that determine whether cSCC stays a skin problem or becomes a systemic one. ✓
“C5aR1 Promotes Invasion, Metastasis, and Poor Prognosis in Cutaneous Squamous Cell Carcinoma.”
— Title, Heiskanen et al., American Journal of Pathology, 2025
A 2018 study in Cancer Cell by Medler and colleagues provided the prior layer of evidence: that C5a is not a bystander in squamous cancer but a co-dominant driver of the process by which normal squamous cells become malignant. In mouse models of squamous carcinogenesis, removing C5aR1 reduced tumor incidence, tumor-associated inflammation, blood vessel formation, and malignant progression to SCC. Human squamous cell carcinomas showed heavy infiltration with C5aR1-positive cells. Patients with low C5aR1 and high cancer-killing T cells had improved survival. ✓
Two independent research groups. Two independent methods. The same target. The same conclusion.
2.3 What the Market Treats
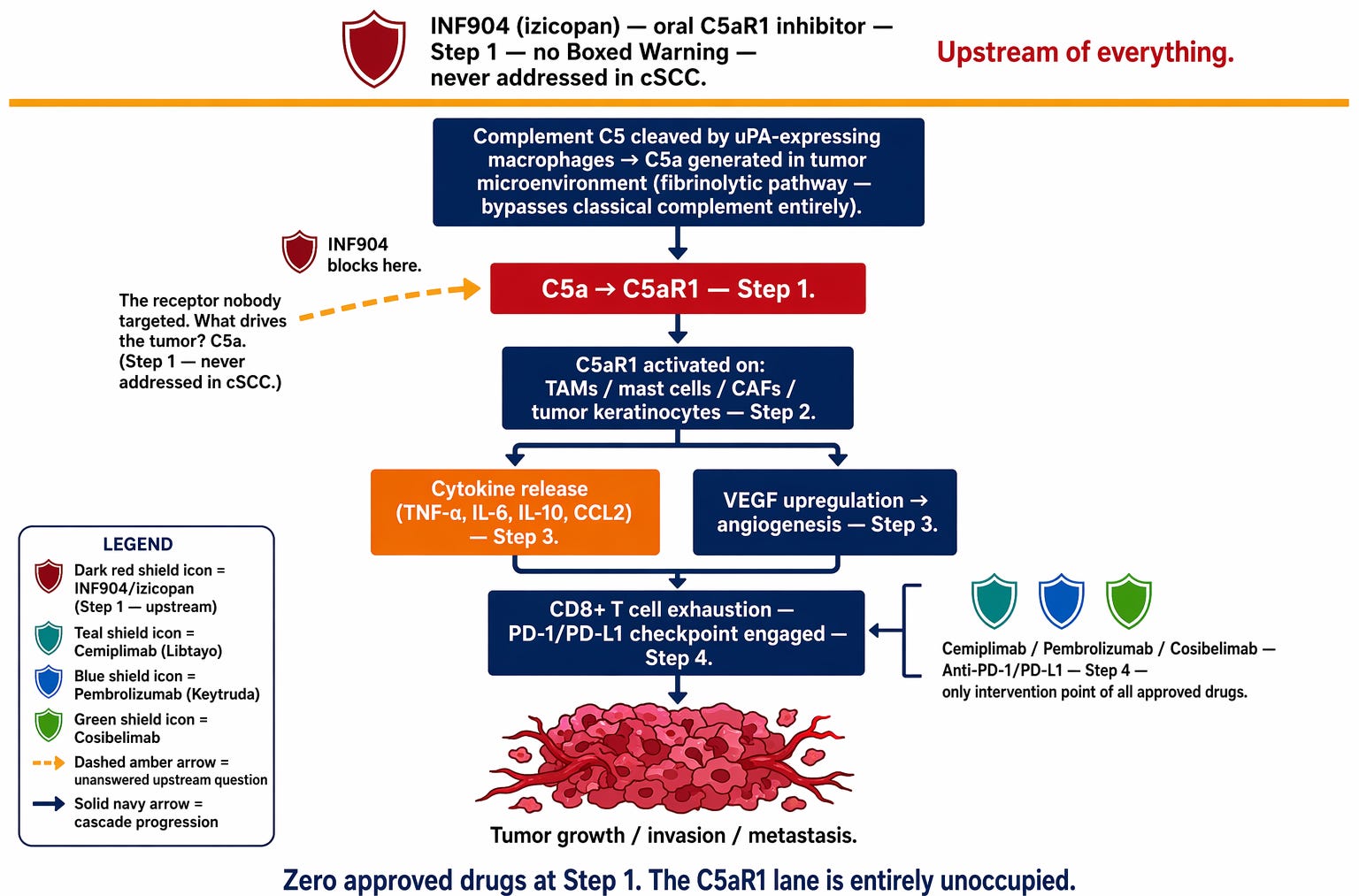
Figure 2a. The C5a cascade in cSCC, read from top to bottom. Above the amber line: INF904, the only intervention at Step 1. Below it: the sequence of events from C5 cleavage through C5a generation, C5aR1 activation, immune suppression, and tumor growth. The three checkpoint inhibitors approved for this disease — cemiplimab, pembrolizumab, cosibelimab — appear at Step 4, the only point in this cascade any approved drug has ever addressed. The lane above them has never had a drug in it.
The conventional understanding of cSCC immunobiology focuses on checkpoint pathways — specifically the PD-1 and PD-L1 proteins. Think of it as a lock-and-key system, just like C5a and C5aR1. PD-L1 is the key, displayed on the surface of cancer cells. PD-1 is the lock, sitting on the surface of T cells. When PD-L1 finds PD-1, the T cell receives a stand-down signal: do not attack. Checkpoint inhibitors block either the key or the lock — the stand-down signal never gets sent, and the T cell remains free to attack.
This is real biology and it produces real clinical benefit. For roughly half of patients with advanced cSCC, unblocking the checkpoint restores enough T-cell function to produce meaningful responses.
But releasing a system-wide immune brake without addressing the upstream driver has consequences. All three approved checkpoint inhibitors carry a Boxed Warning — the FDA’s most serious safety designation, printed at the top of the prescribing label — for immune-mediated adverse reactions: colitis, pneumonitis, liver inflammation, permanent thyroid damage, adrenal insufficiency. These side effects are not incidental. They are the mechanistically inevitable result of disengaging an immune brake without fixing the upstream signal that made that brake necessary in the first place. Gaming the checkpoint without addressing C5a is not fixing what is broken. It is releasing pressure while the tap runs on. ✓/~
Checkpoint inhibitors leave the upstream driver — C5a — entirely unaddressed. The inflammatory program generating the immunosuppressive tumor microenvironment keeps running. For the half of patients who don’t respond — or who respond and then progress — this upstream problem is the reason.
2.4 What the Market Ignores
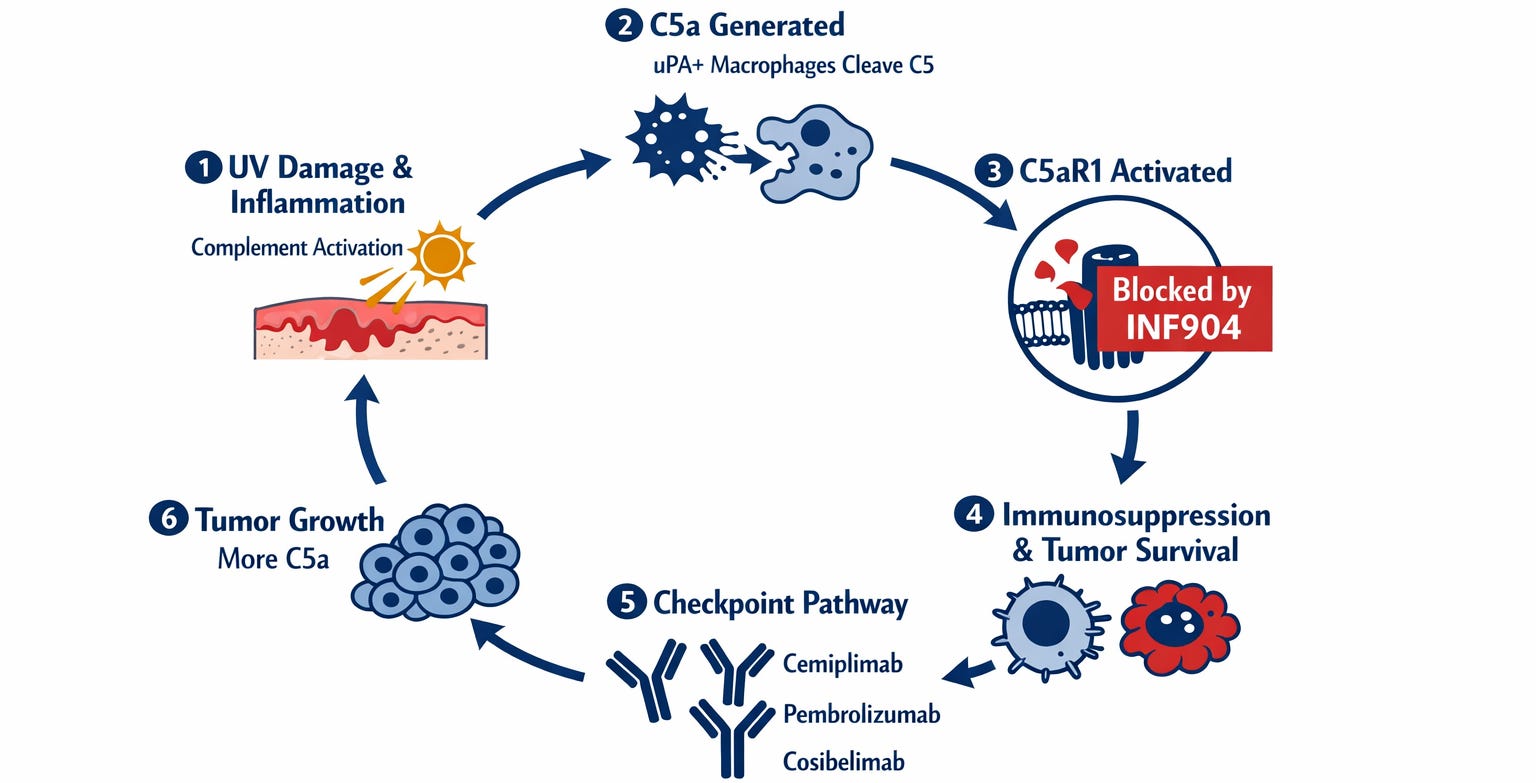
Figure 2b. The C5a signaling cycle in cSCC, shown as a self-sustaining loop. Six stations. Station 1: UV damage initiates complement activation. Station 2: uPA+ macrophages cleave C5, releasing C5a. Station 3: C5aR1 is activated — the point where INF904 intervenes and stops the cycle. Station 4: Immunosuppression, invasion, and intracellular tumor survival in the hypoxic core. Station 5: The checkpoint pathway — the only point any approved drug addresses. Cemiplimab, pembrolizumab, and cosibelimab all intervene here. Station 6: Tumor growth generates more C5a, and the cycle restarts. Without INF904, the cycle continues regardless of what happens at Station 5 — because checkpoint drugs act only at the cell surface, leaving the C5a-driven growth environment inside the tumor fully intact. INF904 intervenes at Station 3. The cycle never starts.
The C5aR1 induction finding
One of the most striking findings in the Heiskanen study: cSCC cells do not express C5aR1 in isolation. The receptor only appears when tumor cells make contact with fibroblasts in the surrounding tissue. ~ The tumor does not arrive with this capability — it acquires it from its own environment. Take away the fibroblast contact, or block the C5aR1 signaling it induces, and the tumor loses the invasive capacity the microenvironment handed it. ✓
C5aR1 on the tumor cell itself — direct apoptosis
C5aR1 is not merely an immune cell receptor in cSCC. The malignant keratinocytes themselves express it. ✓ A 2023 study in the Journal of Clinical Investigation by Beach and colleagues established that blocking C5aR1 in tumor cells shifts the activity of a pro-survival molecular switch called NF-κB toward programmed cell death — specifically in tumor cells, not in normal tissue. ✓ The effect is at least partially independent of T cells. This is not only immune modulation. It is direct tumor cell killing. ✓
Tumor-associated neutrophils and NETosis
The neutrophils present in and around cSCC tumors have been reprogrammed by the C5a-saturated tumor microenvironment into a pro-tumor phenotype called tumor-associated neutrophils (TANs). This distinction matters: in hidradenitis suppurativa, pyoderma gangrenosum, and ANCA-associated vasculitis, neutrophils are the primary effectors — causing direct, catastrophic tissue destruction through NETosis and reactive oxygen species. In cSCC, the damage mechanism is different. TANs suppress cancer-killing CD8+ T cells, promote new blood vessel formation, and release NET-associated proteases that degrade the basement membrane — creating physical corridors for tumor cell invasion. ~
Same cell type. Corrupted function. A neutrophil that should be killing threats is instead dismantling the barriers that contain the tumor — and releasing NETs that activate more complement, generating more C5a, feeding the self-amplifying cycle.
This is why elevated NLR (the neutrophil-to-lymphocyte ratio — a simple measure from a standard blood test that reflects the balance between inflammatory first-responders and cancer-fighting lymphocytes) is clinically meaningful in cSCC. It measures the active TAN-driven immunosuppressive burden in real time. High NLR means the balance has tipped. The upstream signal driving that imbalance is C5a. ✓/~
The hypoxic tumor core — the mechanism no drug has addressed
Solid tumors develop low-oxygen centers. As a cSCC tumor grows faster than its blood supply, the interior becomes severely oxygen-deprived. These hypoxic cores are the most therapy-resistant compartment of any solid tumor — the most immunosuppressive, and the most prognostically dangerous.
In July 2025, a research team at the University of Oxford published a study that changed the mechanistic picture of cSCC — and of every solid tumor with a hypoxic core. ✓
Under severe hypoxia — less than one-tenth of one percent oxygen — cancer cells mount an emergency stress response. Three separate molecular pathways activate simultaneously. All three directly upregulate the expression of C5aR1 in cancer cells. At the same time, hypoxia drives the cancer cell to pull C5aR1 from its surface into its interior via a process called endocytosis. ✓
The cell is producing more C5aR1 — but sending it inside rather than displaying it on the surface. A researcher measuring surface receptor levels would see no change. The survival apparatus being built in the interior would be invisible to them. Total C5aR1 inside the cell increases. Surface expression remains flat. The entire increase is intracellular. ✓
What does intracellular C5aR1 do once inside the cell? It suppresses the cell’s own self-destruction programs — blocking both autophagy (the cellular self-cleaning process) and apoptosis (programmed cell death). ✓ The precise mechanism by which internalized C5aR1 achieves this is not fully established. The most biologically coherent explanation is that it has a ligand to bind inside the cell: C5a generated internally. ~
The autocrine intracellular C5a loop
Tumor cells are not passive recipients of C5a generated by macrophages in the surrounding microenvironment. They generate it themselves, from within — through what the field now calls the complosome (the complement system operating entirely inside the cell, independently of anything circulating in the bloodstream or extracellular space). Cancer cells carry intracellular stores of complement C5 in their own cellular compartments. An enzyme called cathepsin D cleaves this intracellular C5 to generate C5a inside the cell — completely independently of anything happening in the extracellular space. ~ This has been confirmed in cancer cells broadly ✓, with application to cSCC specifically as a supported inference. ~
cSCC tumor cells have additionally been shown to secrete complement proteins C1r and C1s locally in a way that activates downstream complement independently of the normal initiation pathway — direct evidence that cSCC cells generate their own complement activation rather than relying solely on systemic sources. ✓
The implication: intracellular C5aR1 in the hypoxic tumor core may be continuously engaged by C5a the cell is producing for itself, in a self-contained survival circuit that operates entirely behind the plasma membrane, invisible to anything outside the cell. ~ A field consensus review published in February 2026 identified this intracellular C5a/C5aR1 loop in tumor cells as a central mechanism driving immune evasion and therapy resistance across cancer types — and named cell-permeable access to intracellular C5aR1 as the field’s number one therapeutic research priority. ✓
Readers of the ulcerative colitis (UC) article in this series will recognize a parallel worth noting. In UC, the healthy colon is naturally low-oxygen — a carefully maintained anaerobic environment that good bacteria evolved to inhabit. When that environment is disrupted and oxygen floods in, pathogenic bacteria thrive and the disease follows. In cSCC the logic runs in reverse but the principle is the same: a disrupted oxygen environment enables what should not survive. The cancer cell creates its own low-oxygen core and activates survival machinery built for exactly that world — ancient programming running in the wrong context, in the wrong host, at catastrophic cost. The oxygen environment that should signal danger instead signals home.
That ancient programming has a reason to exist. The cell is not broken. It is doing something it was designed to do. The hypoxia survival program almost certainly evolved for contexts where temporary hypoxia is normal and reversible: wound healing, embryonic development, physical exertion. Cancer creates the chronic hypoxic environment that locks this ancient program permanently on. This mechanism was not described in the scientific literature until July 2025. The experiment the science is now pointing toward could not have been conceived before Suwa published. ✓/~
The survival program cancer exploits may be older than we appreciate. The conditions that trigger it — severe cellular oxygen deprivation — resemble the world that existed before the Great Oxidation Event some 2.4 billion years ago, when the Earth’s atmosphere contained no free oxygen and cells built their survival machinery around hypoxia as the default state of existence. The survival switch cancer hijacks may be older than oxygen itself. ~
Why antibodies cannot reach it
Monoclonal antibodies — the drug class that includes cemiplimab, pembrolizumab, and vilobelimab — are large molecules. Their molecular weight is approximately 150,000 daltons, roughly one thousand times the size of a common anti-inflammatory tablet. They cannot cross the plasma membrane of a living cell. ✓ C5a itself — a small peptide — also cannot. Both are pharmacologically blind to intracellular C5aR1. The Oxford research team stated this explicitly and named two active oncology clinical trials using anti-C5aR1 antibodies, cautioning that their limited membrane permeability would constrain efficacy against intracellular receptor pools. ✓
The implication is direct: every anti-C5a antibody — including vilobelimab — can only address C5aR1 on the cell surface. The survival program in the hypoxic tumor core, driven by internalized intracellular C5aR1 engaged by autocrine intracellular C5a, runs on untouched.
This raises an immediate question: if intracellular C5aR1 is the key survival mechanism in the hypoxic tumor core, and vilobelimab cannot reach it, why did the 79-year-old patient achieve a biopsy-confirmed complete remission on vilobelimab?
The answer is that vilobelimab does address the extracellular C5a-driven immunosuppressive tumor microenvironment — and for one patient in ten, addressing the surface mechanism alone was sufficient to produce a response. Neutralizing circulating C5a partially restores CD8+ T-cell function, reduces macrophage-driven immunosuppression, and removes the invasion signal at the tumor surface. In a patient whose tumor architecture had a less-established hypoxic core, that partial mechanism was enough. ✓/~
The 10% complete remission rate is not a contradiction of the intracellular thesis — it is the evidence that C5a blockade works in this disease. The 90% who did not achieve complete remission are the evidence that surface-only blockade is insufficient. The intracellular mechanism explains both numbers simultaneously. A cell-permeable small molecule that addresses all of it — surface immunosuppression, direct tumor cell apoptosis, TAN reprogramming, and intracellular survival in the hypoxic core — is a categorically different experiment. That experiment has never been run. ~
The drug class that works
The Oxford team tested three C5aR1 inhibitors in hypoxic tumor cell models.✓
PMX205, which has poor cell permeability, produced minimal effect on cancer cell viability under hypoxia and no measurable effect on tumor spheroid growth. ✓
Avacopan and JPE-1375 — both cell-permeable small molecules — significantly reduced cancer cell survival under hypoxia, increased programmed cell death, and suppressed tumor spheroid growth specifically in the low-oxygen regions of the tumor model. After five days of avacopan treatment, tumor spheroid growth was measurably suppressed. ✓
The key variable was not which receptor was targeted. It was whether the drug could get inside the cell to reach it.
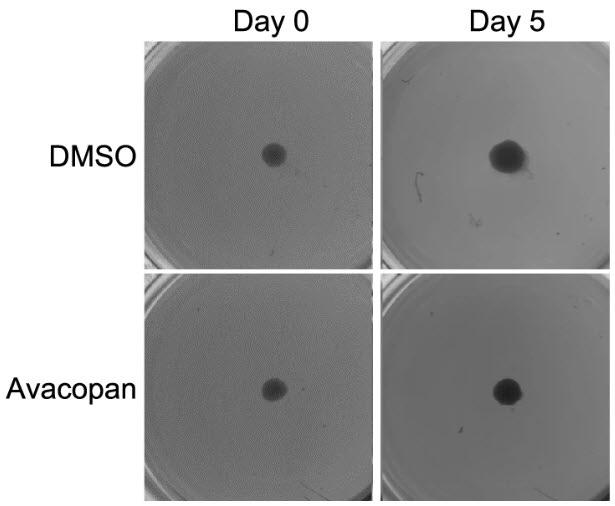
Figure 2c. Two miniature tumor spheroids — three-dimensional clusters of cancer cells grown in a laboratory dish — photographed at Day 0 and Day 5. Top row: the control spheroid has grown visibly larger over five days. Bottom row: the spheroid treated with a cell-permeable C5aR1 inhibitor has not. Same cells. Same conditions. One variable: a drug that can follow C5aR1 inside. Source: Suwa et al., Cell Death & Disease, 2025 (CC BY 4.0).
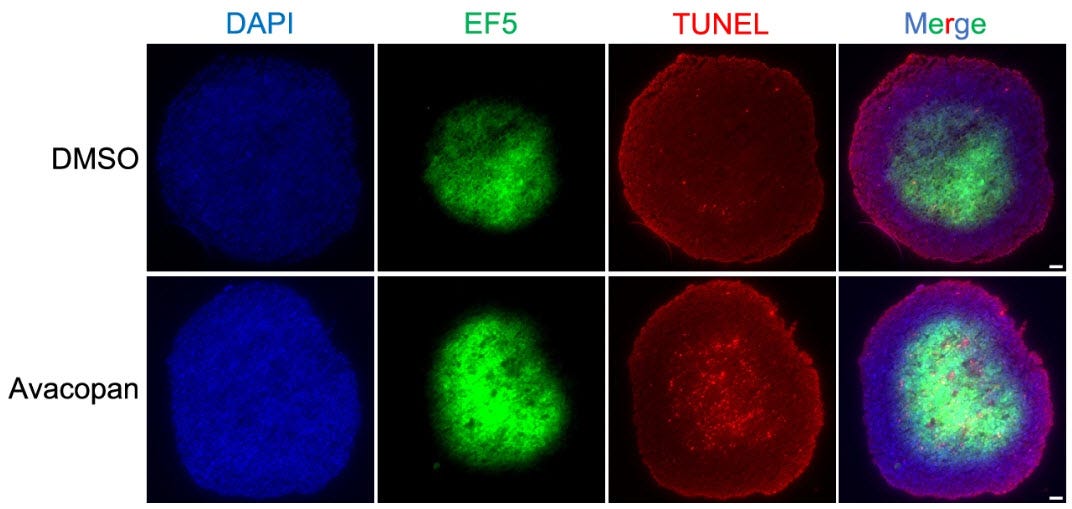
Figure 2d. The same tumor spheroids cut open and stained. Blue marks all cell nuclei. Green marks oxygen-rich regions. Red marks cells undergoing programmed death. Top row (control): less green, sparse red — low oxygen regions are keeping cancer cells alive, protected by intracellular C5aR1. Bottom row (treated with a cell-permeable C5aR1 inhibitor): more green, red flooding the same regions — those same cells are now dying. The outer ring remains largely unaffected in both rows — confirming the drug is not indiscriminately toxic but is selectively killing the cells that hypoxia was protecting. The drug found the tumor's most protected compartment. Every antibody approved for this disease cannot. Source: Suwa et al., Cell Death & Disease, 2025 (CC BY 4.0).
2.5 C5aR1 Is Upstream of Everything in cSCC
Blocking C5aR1 with a cell-permeable small molecule simultaneously addresses five distinct pro-tumor mechanisms — none of which any approved drug in cSCC touches. ~
First, the immunosuppressive tumor microenvironment. Blocking C5aR1 on macrophages and other immunosuppressive myeloid cells restores CD8+ T-cell function. In the Medler study, C5aR1 blockade combined with chemotherapy converted exhausted T cells into functional cancer-killing cells. ✓
Second, tumor cell invasion. Blocking C5aR1 on malignant keratinocytes removes the invasion signal C5a provides. C5a drove invasion through collagen at p < 0.0001 in the Heiskanen study. ✓
Third, tumor cell survival in the hypoxic core. Blocking intracellular C5aR1 removes the survival program keeping the most therapy-resistant cells alive. Cell-permeable small molecules kill hypoxic tumor cells. Non-permeable compounds and antibodies do not. ✓
Fourth, angiogenesis — the formation of new blood vessels feeding tumor growth. Blocking C5aR1 on macrophages and mast cells reduces VEGF signaling and vessel formation. Anti-VEGF drugs exist in oncology — bevacizumab, for example — and target exactly this downstream consequence of C5a signaling. They have not established meaningful efficacy in cSCC. Mopping one downstream signal while the tap runs on. INF904 fixing the tap suppresses VEGF upregulation as one of five simultaneous mechanisms. ✓/~
Fifth, TAN-driven basement membrane degradation. Blocking C5aR1 at the source reduces the signal that reprograms neutrophils into their pro-tumor TAN phenotype — reducing NET release, protease activity, and the physical degradation of the basement membrane that enables invasion. Cut the signal that corrupts the neutrophil and the neutrophil returns to doing what it evolved to do. ~
2.6 Extension Inference — Head and Neck Squamous Cell Carcinoma
Head and neck squamous cell carcinoma (HNSCC) shares the same squamous cell histology as cSCC, the same C5a-driven carcinogenesis mechanism, and the same tumor microenvironment immunosuppression pattern. The same hypoxia-driven intracellular C5aR1 survival mechanism established by Suwa applies to all squamous tumors with hypoxic cores — and HNSCC tumors, by their anatomy, are frequently hypoxic. A 2025 systematic review of complement signaling in cancer confirms C5a’s role in HNSCC specifically. ~
If INF904 produces clinical responses in checkpoint non-responders with cSCC via intracellular C5aR1 blockade, the first extension indication is HNSCC — approximately 67,000 new US cases per year, with worse outcomes than cSCC and higher unmet need. ? That is a different article. But the biology points there directly.
3: What Exists Today
Three drugs are approved for advanced cSCC. All three are checkpoint inhibitors. All three carry a Boxed Warning for immune-mediated adverse reactions. All three address the same step in the same pathway. Each earns the revenue it generates.
Cemiplimab (Libtayo — Regeneron/Sanofi)
Cemiplimab was the first systemic therapy approved for advanced cSCC, receiving FDA approval in 2018. Before it, patients with locally advanced or metastatic disease had no approved treatment. ✓ The approval was based on an overall response rate of approximately 47% in the EMPOWER-CSCC-1 trial, with durable responses in those who responded. ✓ For nearly half of patients with a disease that previously had no options, cemiplimab is a genuine advance. It generates over one billion dollars annually across its approved indications.
At approximately $190,000 to $200,000 per patient per year at US list price, cemiplimab is among the most expensive drugs in oncology. ~ That cost is delivered via intravenous infusion at an infusion center every three weeks — a meaningful burden for an elderly patient population. Its Boxed Warning covers immune-mediated reactions that are the direct consequence of releasing a system-wide immune brake without addressing the upstream driver.
What it does not do: address the C5a-driven inflammatory program generating the immunosuppressive tumor microenvironment. Reach intracellular C5aR1 in the hypoxic tumor core. The approximately 53% of patients who do not respond are left without a mechanistically distinct alternative.
Pembrolizumab (Keytruda — Merck)
Pembrolizumab received FDA approval for recurrent or metastatic cSCC not curable by surgery or radiation in 2020. ✓ Response rates are approximately 35%. ✓ Pricing is comparable to cemiplimab — approximately $180,000 to $200,000 per year. ✓ The mechanistic limitation is identical: PD-1 blockade downstream of C5a-driven immune suppression. Boxed Warning: immune-mediated reactions. The approximately 65% of patients who do not respond face the same unmet need.
Cosibelimab (Unloxcyt — Sun Pharma, acquired from Checkpoint Therapeutics March 2025)
Cosibelimab received FDA approval in December 2024, targeting PD-L1 rather than PD-1. ✓ To extend the lock-and-key analogy: PD-L1 is the key the cancer cell displays; PD-1 is the lock on the T cell. Cemiplimab and pembrolizumab block the lock. Cosibelimab blocks the key. Different point of intervention — same stand-down signal interrupted. ✓ Response rates are approximately 47%. ✓ Cosibelimab also demonstrates antibody-dependent cellular cytotoxicity (ADCC) — an additional mechanism by which it can direct immune effector cells to kill tumor cells directly — though the clinical contribution of this feature relative to PD-L1 blockade alone remains to be fully characterized. ✓/~
Pricing is in the same range as the other checkpoint inhibitors — approximately $220,000 per year at estimated list price. ~ Boxed Warning: immune-mediated reactions. Mechanistic limitations identical to the other agents in the class.
The Market Signal
The global cSCC treatment market was valued at approximately $13.7 billion in 2024 and is projected to reach $27.5 billion by 2034 — a market roughly doubling in size over the decade. ~ The combined revenue of the checkpoint inhibitors across their oncology indications exceeds tens of billions annually. That revenue is real and deserved — it is the revenue of drugs that work for roughly half of patients in a disease that previously had no options. ~
The other half — the 50 to 65% who do not respond to checkpoint inhibitors, or who respond and then progress — represent a market of approximately equal size with no approved second-line option addressing a distinct mechanism. ~ At $180,000 to $220,000 per year, those patients pay the same price, endure the same Boxed Warning side effect risk, and receive no benefit.
INF904 is an oral pill taken at home. No infusion center. No IV administration costs. No Boxed Warning. No immunogenicity risk that comes with antibody-based therapy. Priced at an assumed $75,000 to $100,000 per year — roughly half the cost of the checkpoint inhibitors it would follow — for a population with no approved alternative. ~ In a disease where the average patient is 74 years old, the difference between driving to an infusion center every three weeks and taking a pill daily at home is not a minor convenience. It is a quality of life difference that affects treatment adherence, caregiver burden, and real-world effectiveness.
For a payer currently spending $200,000 per year on a drug that fails two-thirds of patients in the second-line setting, a more targeted oral drug addressing a distinct upstream mechanism at half the cost is not a difficult value argument. ~
The Competitive Landscape at a Glance
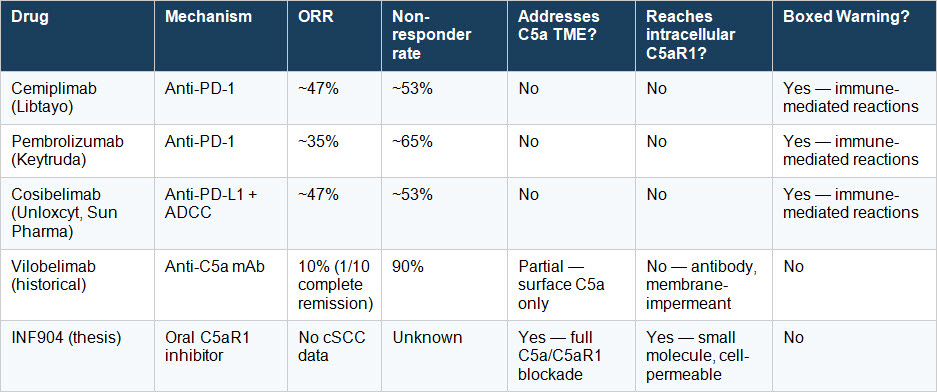
The vilobelimab row is historical — it establishes clinical proof that C5a blockade produces a response in this disease. The INF904 row is a thesis. No cSCC data exists. The scientific argument for why it should work is the subject of this article. ~/?
4: The Patient’s Journey
The diagnostic journey in cSCC is not the multi-year odyssey of misdiagnosis that characterizes hidradenitis suppurativa or pyoderma gangrenosum. cSCC is usually diagnosed correctly. The lesion is visible. Dermatologists know what to look for. A biopsy confirms it.
The odyssey in cSCC is different. It is the journey from ‘this is a manageable skin problem’ to ‘this is going to kill me.’
The typical patient with advanced cSCC is elderly — the mean age in the vilobelimab trial was 79 years. ✓ The lesion is usually on sun-damaged skin: the scalp, the face, the ears, the back of the hands. It may have been there for years, treated conservatively, biopsied, excised, declared clear. And then it comes back.
At that point, the standard pathway is systemic therapy. Cemiplimab or pembrolizumab. Approximately half of patients respond. For those who do not, or who respond and then progress, the options contract sharply. Platinum-based chemotherapy can provide modest benefit in some patients, but carries significant toxicity in an elderly population. Clinical trials, if available. Nothing else with a distinct mechanism.
The Complement Signal Hiding in Plain Sight
Every patient whose cSCC tumor shows high C5aR1 expression has an elevated metastatic risk. ✓ The Heiskanen study established this with p < 0.0001 statistical precision across two independent patient groups. C5aR1 on tumor cells predicts who dies. C5aR1 in the surrounding fibroblasts predicts who dies. Both markers, independently.
No oncologist today routinely orders a C5aR1 stain when staging cSCC. The test exists. The published data on what it predicts exists. The therapeutic target it points to has no approved drug. The signal is sitting in the pathology lab, unread.
C5aR1 immunohistochemistry tells you the most — it identifies the tumor biology directly and predicts who dies. But it requires archival tissue and a staining protocol not yet in routine clinical practice. While the field works toward making C5aR1 tissue testing routine, there is already a simpler proxy available from a standard blood draw that identifies the same high-risk population prospectively, before treatment begins.
The NLR Proxy
The neutrophil-to-lymphocyte ratio — the NLR — is a standard measure derived from a complete blood count (CBC), the most basic blood test in medicine. It reflects the balance between the immune system’s inflammatory first-responders (neutrophils) and its adaptive cancer-fighting arm (lymphocytes). When C5a chronically activates neutrophils and reprograms them into the pro-tumor TAN phenotype, the ratio shifts. The NLR rises.
A 2025 study by Xiao and colleagues examined 139 cSCC patients receiving checkpoint immunotherapy. ✓ NLR was the strongest single predictor of non-response, with an area under the curve — a measure of predictive accuracy where 1.0 is perfect and 0.5 is chance — of 0.775. A combined model incorporating NLR alongside other blood markers reached 0.878 with 93% specificity. ✓
Elevated NLR does not merely correlate with checkpoint failure — it measures the active TAN-driven immunosuppressive burden that explains it. High NLR means the tumor microenvironment is heavily polarized toward innate immune dominance. The checkpoint inhibitor frees the T cell at one gate while TANs are blocking it at several others. Blocking C5aR1 addresses the upstream signal that created and maintains that polarization. ✓/~
The patients least likely to benefit from the only approved treatments are identifiable today, with a standard blood draw, before treatment begins. ✓ They are not being systematically identified in clinical practice for the purpose of directing them toward a mechanistically distinct intervention. Because no mechanistically distinct intervention has ever been tested in this population. Yet.
Section 5: The Patient Population
All commercial calculations are US-only unless noted. All population estimates are labeled ~ and reflect published literature and company estimates.
Approximately one million Americans are diagnosed with cSCC each year. ✓ The vast majority have early-stage disease treated successfully with surgery — though the true incidence is likely higher than reported, given how frequently early lesions go unexamined on sun-damaged skin in elderly patients. ~
Of those million annual cases, approximately 15,000 to 20,000 develop locally advanced disease not amenable to surgery, and a further 7,000 to 10,000 develop metastatic disease. Together, the advanced cSCC population is approximately 25,000 to 30,000 per year on conservative estimates, and up to 45,000 on corrected published estimates that account for the full locally advanced plus metastatic population. ~
Of those with advanced disease, approximately 70% receive systemic therapy — approximately 31,500 patients per year on the corrected estimate. ~
Of those receiving systemic therapy, approximately 55% are checkpoint inhibitor non-responders or progressors — approximately 17,000 patients per year. ~
That is the INF904 patient. Approximately 17,000 Americans per year with advanced cSCC who have already failed the best available therapy and have nowhere else to go. ~
The European Union and United Kingdom together represent approximately 1.5 to 2 times the US volume in advanced cSCC. Japan represents approximately 0.3 times the US. All peak sales calculations in Section 8 are US-only, with ex-US totals noted separately.
Section 6: Finding the Right Patient
Primary enrollment biomarker: NLR
NLR directly measures the neutrophil hyperactivation and TAN-driven immune imbalance that C5a drives. A patient with elevated NLR has elevated C5a-driven inflammatory burden — the biology INF904 is designed to address. NLR also prospectively identifies the non-responder population before checkpoint therapy begins. And NLR is a routine blood test requiring no specialty assay. ~
Serum C5a is not appropriate as a patient selection criterion. C5a is chemically unstable in blood samples and fluctuates with collection conditions. InflaRx has used it only as a pharmacodynamic readout to confirm drug effect. ✓
Secondary biomarker: C5aR1 immunohistochemistry
C5aR1 immunohistochemistry on archival tumor samples could serve as an exploratory biomarker, confirming that enrolled patients have the on-target biology INF904 is designed to address. It is not appropriate as an enrollment criterion in a first study — it requires archival tissue and standardized scoring methods not yet validated for prospective enrollment. It belongs in the exploratory endpoint stack. ~
H3.1 nucleosomes as a pharmacodynamic marker
H3.1 nucleosomes are a measurable blood marker for NETosis — the process by which activated TANs release their internal DNA as sticky, web-like neutrophil extracellular traps (NETs). H3.1 nucleosome levels, measurable via the Nu.Q® NETs assay developed by Volition (VNRX), are elevated across multiple neutrophil-driven inflammatory diseases and correlate with disease severity. ~ In an INF904 cSCC trial, falling H3.1 nucleosome levels would confirm that the drug is reducing NETosis activity and the TAN-driven basement membrane degradation described in Section 2.4. This would be a secondary pharmacodynamic endpoint, not an enrollment criterion.
Enrollment strategy
InflaRx already identified the right patient in the vilobelimab trial: advanced cSCC with documented progression on at least one prior checkpoint inhibitor, PD-1/PD-L1 therapy as the most recent treatment, ECOG performance status (a standardized measure of patient functional fitness, where 0 is fully active and 5 is deceased) of ≤1, and accessible lesions for biopsy. An INF904 Phase 2a starts from the same population definition — with NLR ≥ 3.5 at screening added as the enrollment criterion that identifies the mechanistically appropriate patient. ~
Section 7: What a Study Would Need to Show
INF904 is InflaRx’s oral, selective small molecule C5aR1 inhibitor. In Phase 1 single ascending dose studies, INF904 demonstrated approximately three-fold higher peak drug levels, approximately ten-fold higher total drug exposure, and doubled in vivo inhibitory effect compared to avacopan — the only approved C5aR1 inhibitor — at comparable doses. ✓ INF904 carries no Boxed Warning. Its clean safety profile has been established across approximately 200 patients across multiple indications. ✓ As a small molecule, it carries no risk of anti-drug antibody development that comes with antibody-based therapy.
Critically for cSCC: INF904 is cell-permeable. Avacopan is also a cell-permeable small molecule — and avacopan, in the Oxford hypoxic tumor model, killed hypoxic tumor cells that antibodies and peptides could not reach. ✓ INF904’s approximately ten-fold higher total drug exposure versus avacopan at a comparable dose means it delivers that cell-permeable activity at significantly greater intensity. ~ The experiment suggested by the Oxford data has never been run with INF904 — or with any C5aR1 inhibitor in a patient with advanced cSCC. This is the study.
Phase 2a — Proof of Concept
Design: Single-arm, open-label. N = 20 to 30 patients. Advanced cSCC with documented progression on at least one prior checkpoint inhibitor. NLR ≥ 3.5 at screening. ECOG PS ≤ 1. INF904 at the dose established in Phase 1 multiple ascending dose studies. Duration: 12 weeks for primary assessment, continued dosing through 24 weeks for patients with confirmed response. Mandatory paired biopsies at baseline and on-treatment at week 4, consistent with the biopsy protocol InflaRx established in the vilobelimab cSCC trial. ~
Primary endpoint: Best Overall Response Rate (Best ORR) — confirmed complete remission or partial response by both standard imaging criteria (RECIST v1.1) and immune-modified criteria (iRECIST). The dual assessment is used because immune-mediated responses can show initial apparent progression before responding — a patient who looks like they are worsening early may be experiencing immune activation, not treatment failure. This framework was established by InflaRx in the vilobelimab cSCC trial. ~
Key secondary endpoints: Disease control rate (complete remission + partial response + stable disease) at 12 weeks. Progression-free survival. Overall survival at the 24-week landmark. NLR change from baseline at 12 weeks. Serum C5a change from baseline. H3.1 nucleosome change from baseline. C5aR1 immunohistochemistry on paired biopsies at baseline versus on-treatment at week 4 — directly confirming intracellular C5aR1 reduction. Patient-reported quality of life using a validated scale — particularly meaningful in an elderly patient population where an oral drug at home represents a meaningfully different experience from IV infusion every three weeks. Safety and tolerability. ~
What Phase 2a needs to show: an ORR of at least 20 to 25% in the checkpoint failure population, with NLR normalization in at least 50% of patients, would constitute sufficient proof of concept to advance. Thresholds to be confirmed with biostatistical input before study initiation. ~
Phase 2b does not enroll simultaneously with Phase 2a. The two-stage design is consecutive. Phase 2b protocol, sample size, and endpoints are informed by Phase 2a data. IND preparation and site activation planning can run in parallel with Phase 2a enrollment, but patient enrollment in Phase 2b waits for Phase 2a results.
Phase 2b — Efficacy
Design: Randomized, controlled. N = 80 to 120 patients. Advanced cSCC, checkpoint inhibitor failure, NLR ≥ 3.5, ECOG PS ≤ 1. INF904 at the Phase 2a-confirmed dose versus placebo or investigator’s choice second-line therapy. Primary endpoint: progression-free survival at 24 weeks. ~
Phase 3 and Regulatory Path
If Phase 2b produces a meaningful progression-free survival signal, FDA Breakthrough Therapy Designation may be sought — a status enabling more frequent FDA guidance and rolling review. In advanced cSCC with checkpoint inhibitor failure, median overall survival is less than 12 months and no approved second-line agent with a distinct mechanism exists. The unmet need qualification is clear. ~
An accelerated approval pathway based on overall response rate as a surrogate endpoint is plausible if Phase 2b response rates in the NLR-high checkpoint failure population reach 30% or above with durable responses. Confirmatory overall survival data would follow. ~
There is a constraint that must be stated plainly: InflaRx cannot self-fund a Phase 3 oncology program at current resource levels. Any development beyond a Phase 2a in cSCC requires a partner with oncology commercial infrastructure. This is a thesis article. The thesis requires a decision to run the experiment — and that decision is not InflaRx’s alone to make. ?
Section 8: Peak Sales Estimate
All estimates are US-only unless noted. All figures are labeled ~ as they involve population modeling and penetration assumptions. Pricing is assumed at $75,000 to $100,000 per year — the pricing framework applied consistently across this series for oral small molecule C5aR1 inhibitors, reflecting approximately half the cost of the checkpoint inhibitors this drug would follow. ~
Market Context
The global cSCC treatment market was valued at approximately $13.7 billion in 2024 and is projected to reach $27.5 billion by 2034 — a market roughly doubling in size over the decade, driven by an aging population, rising incidence, and expanding treatment options. ~ Within that growing market, the checkpoint non-responder segment — INF904’s specific opportunity — currently generates substantial checkpoint drug revenue for patients who receive no benefit. That segment has no approved second-line option with a distinct mechanism. ~
Patient Population Funnel (US only)
Advanced cSCC reaching systemic therapy: approximately 31,500 per year. ~
Checkpoint inhibitor non-responders at 55%: approximately 17,000 per year. ~
Reach second-line systemic evaluation and are candidates for an oral agent (approximately 60%): approximately 10,200. ~
Addressable with NLR-elevated enrollment criterion (approximately 70% of the above): approximately 7,140. ~
The INF904 patient: approximately 7,000 US patients per year in the near-term addressable population. ~
Three Scenarios
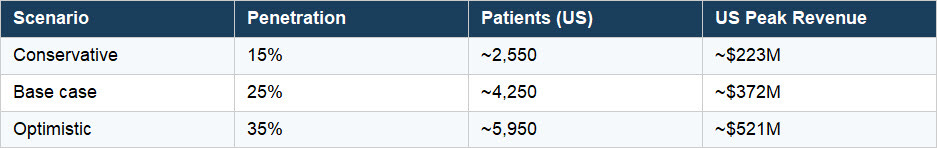
Ex-US markets (EU + Japan) are estimated at approximately 1.5 times the US peak revenue figure across all three scenarios. ~
The Value Proposition
At $75,000 to $100,000 per year assumed — roughly half the cost of the checkpoint inhibitors it would follow — INF904 enters as an oral pill with no infusion center requirement, no IV administration burden, no Boxed Warning, no immunogenicity risk, and a mechanism that addresses what checkpoint therapy leaves completely untouched. For a payer currently spending $200,000 per year on a drug that fails two-thirds of patients in the second-line setting, this is not a difficult value argument. ~
The Bigger Picture
These numbers are modest by large-cap oncology standards. They are not the commercial thesis.
The cSCC program is a proof-of-concept thesis. If INF904 produces a meaningful response rate in checkpoint non-responders by addressing intracellular C5aR1 in the hypoxic tumor core, the implication is not a $500 million cSCC drug. The implication is that every solid tumor with a hypoxic core — and most solid tumors do have one — becomes a potential indication for a cell-permeable C5aR1 inhibitor. ~
Head and neck squamous cell carcinoma: approximately 67,000 new US cases per year. Lung squamous cell carcinoma: approximately 50,000 new US cases per year. Bladder SCC variants. Esophageal SCC — where the Heiskanen TCGA analysis found elevated C5AR1 correlates with shorter overall survival. ✓
The cSCC program is the clinical proof that unlocks the class. ? The peak sales estimate above is the floor. What comes after depends on whether the experiment is run.
Section 9: Why It Matters
The Science
Four independent research groups, across three countries, over seven years, have arrived at the same place. ✓
Medler and colleagues established in 2018 that C5a is a co-dominant driver of squamous carcinogenesis and that blocking C5aR1 reduces tumor incidence, inflammation, blood vessel formation, and malignant progression. Beach and colleagues established in 2023 that malignant epithelial cells express C5aR1 themselves, and that blocking it triggers programmed cell death specifically in tumor cells. Heiskanen and colleagues established in 2025 that C5aR1 expression in human cSCC tissue tracks metastatic risk and independently predicts who dies — at p < 0.0001. Suwa and colleagues established in 2025 that solid tumors under hypoxia upregulate and internalize C5aR1 as a survival mechanism, that cell-permeable small molecules can reach and block intracellular C5aR1, and that antibodies and peptides cannot. ✓
One patient — 79 years old, Stage IV, failed the best drug the field had available — received an anti-C5a antibody. Her tumor disappeared. The response lasted 512 days. The antibody was the wrong tool class for the deepest mechanism. She responded anyway. ✓
What has not been done: no one has given a cell-permeable oral C5aR1 inhibitor to a patient with advanced cSCC. The experiment that the science has been building toward for seven years has never been run. ✓/?
The survival program cancer exploits in the hypoxic tumor core has roots deeper than the disease itself. Before the Great Oxidation Event some 2.4 billion years ago, the Earth contained no free oxygen. Cells built their survival machinery around hypoxia as the default state of existence. The switch that cancer pulls — hiding C5aR1 inside itself to escape the oxygen-dependent world outside — may be a remnant of machinery built for a world before oxygen existed. The survival switch cancer hijacks may be older than oxygen itself. ~ What we know for certain is that it was not described in the scientific literature until 2025 — and that the experiment it implies has not yet been run. ✓/?
The Patient Face
That market is made of people.
The 50 to 65% of advanced cSCC patients who do not respond to checkpoint therapy are not a non-responder rate. They are elderly patients — mean age 74 in the vilobelimab trial — with tumors on their scalp, their face, their ears. Tumors that bleed and do not heal. Tumors that have already been treated with surgery, with radiation, with the best immunotherapy medicine currently offers, and that kept growing anyway.
For those patients, there is no approved second-line option with a distinct mechanism. There is no drug that goes where the tumor has retreated — into the hypoxic core, behind the plasma membrane, beyond the reach of antibodies. There is no standard blood test being used systematically to identify them before treatment begins and direct them toward something different.
That is not inevitable. It is a development priority that has not yet been acted on.
The Urgency
INF904 is in active development. InflaRx is finalizing the study design for an INF904 Phase 2b in hidradenitis suppurativa. An AAV program is under consideration but has not been confirmed, with advancement contingent on capital and partnership progress. Capital is constrained. A cSCC Phase 2a — in checkpoint non-responders, with an NLR enrollment criterion and a paired-biopsy pharmacodynamic endpoint — would require a partner or non-dilutive funding. That conversation has not happened publicly. ✓
But the science is ready. The patient population is identifiable — today, with a routine blood draw. The biological rationale is peer-reviewed and independent of any single research group or pharmaceutical company. The drug exists. The only missing ingredient is the decision to run the experiment.
A 79-year-old woman with a scalp tumor that had failed cemiplimab told us something important in 2022 and 2023. It took until 2025 for a research group in Oxford to explain why her tumor responded — and why the next version of that experiment, with a drug that follows C5aR1 all the way inside, might produce a different ending for the patients who are still waiting. Science moves. Capital follows. The question is how long the gap stays open.
If you know someone living with an advanced skin cancer that has stopped responding to treatment — a lesion that bleeds and will not heal, a diagnosis that came back after surgery, a tumor the best available drug could not stop — this article was written with them in mind. The science that could change their options exists. It is published. It is peer-reviewed. It is pointing at a target that no approved drug has yet addressed. Share this article with the people who need to know that the biology is ready. That is not inevitable. It is a development priority that has not yet been acted on.
Sources & Further Reading
Sections 2 & 3 — Biology and Competitive Landscape
Heiskanen L, Nissinen L, Siljamäki E, Knuutila JS, Pellinen T, Kallajoki M, Heino J, Riihilä P, Kähäri VM. C5aR1 Promotes Invasion, Metastasis, and Poor Prognosis in Cutaneous Squamous Cell Carcinoma. Am J Pathol. 2025;195(6):1158–1171. doi: 10.1016/j.ajpath.2025.02.004. PMID: 40056975. PMCID: PMC12163391. Primary human cSCC pathology anchor. 174 invasive cSCCs plus staged tissue series from normal skin through metastases. C5aR1 tracks metastatic risk stepwise. Disease-specific 5-year survival (p<0.0001 log-rank, both tumor cell surface and CAF C5aR1 as independent predictors). C5a drives invasion through collagen I in 3D spheroid model (p<0.0001 at 96h). C5aR1 expression induced by fibroblast co-culture at invasive edge. TCGA: elevated C5AR1 correlates with poor OS in esophageal and lung SCC. Used in Sections 2.2, 2.4, 2.5, and 4. https://ajp.amjpathol.org/article/S0002-9440(25)00073-2/fulltext
Suwa T, Lee KSW, Chai IJ, Clark HOL, MacLean DJ, Machado N, Rodriguez-Berriguete G, Singh L, Higgins GS, Hammond EM, Olcina MM. UPR-induced intracellular C5aR1 promotes adaptation to the hypoxic tumour microenvironment. Cell Death Dis. 2025;16:547. doi: 10.1038/s41419-025-07862-z. PMID: 40695778. PMCID: PMC12284258. Published 22 July 2025. Open access. CC BY 4.0. Establishes the intracellular C5aR1 survival mechanism under hypoxia. UPR-dependent C5aR1 upregulation and endocytosis-driven receptor internalization. Intracellular C5aR1 suppresses autophagy and apoptosis. Cell-permeable small molecules (avacopan, JPE-1375) kill hypoxic tumor cells; non-permeable compounds and antibodies do not. Authors explicitly caution against antibody-based C5aR1 targeting in oncology. Source for Figures 2c and 2d (CC BY 4.0). Used in Sections 2.4 and 7. https://www.nature.com/articles/s41419-025-07862-z
Medler TR, Murugan D, Horton W, Kumar S, Cotechini T, Forsyth AM, et al. Complement C5a fosters squamous carcinogenesis and limits T cell response to chemotherapy. Cancer Cell. 2018;34(4):561–578.e6. doi: 10.1016/j.ccell.2018.09.003. PMID: 30300579. PMCID: PMC6246036. Establishes C5a as co-dominant driver of squamous carcinogenesis. uPA+ macrophages generate C5a via C3-independent fibrinolytic pathway. C5aR1 knockout reduces tumor incidence, inflammation, angiogenesis, and malignant progression. C5aR1 blockade plus chemotherapy restores CD8+ T-cell function. Used in Sections 2.2, 2.4, 2.5, and 2.6. https://www.cell.com/cancer-cell/fulltext/S1535-6108(18)30417-3
Beach C, MacLean D, Majorova D, Melemenidis S, Nambiar DK, Kim RK, et al. Improving radiotherapy in immunosuppressive microenvironments by targeting complement receptor C5aR1. J Clin Invest. 2023;133(23):e168277. doi: 10.1172/JCI168277. PMID: 37824211. PMCID: PMC10688992. C5aR1 expressed on malignant epithelial cells; C5aR1 blockade triggers NF-κB-dependent apoptosis specifically in tumor cells, not normal tissue; effect partially T-cell independent. PMX205 (poor cell permeability) does not suppress tumor growth in vivo. Used in Section 2.4. https://www.jci.org/articles/view/168277
Riihilä P, Viiklepp K, Nissinen L, Farshchian M, Kallajoki M, Kivisaari A, Meri S, Peltonen J, Peltonen S, Kähäri VM. Tumour-cell-derived complement components C1r and C1s promote growth of cutaneous squamous cell carcinoma. Br J Dermatol. 2020;182(3):658–670. doi: 10.1111/bjd.18095. PMID: 31049937. PMCID: PMC7065064. Primary source for cSCC-specific complement generation finding. cSCC tumor cells express and secrete C1r and C1s in a C1q-independent manner, activating downstream complement and promoting tumor growth and vascularization. Knockdown of C1r and C1s promoted apoptosis and suppressed xenograft tumor growth in vivo. Used in Section 2.4. https://academic.oup.com/bjd/article/182/3/658/6731441
Oliveira-Tore CF, Giaretta de Moraes A, Plácido HMBS, Signorini NMDL, Fontana PD, Batista Godoy TdP, Boldt ABW, de Messias I. Non-canonical extracellular complement pathways and the complosome paradigm in cancer: a scoping review. Front Immunol. 2025;16:1519465. doi: 10.3389/fimmu.2025.1519465. PMID: 40370471. eCollection 2025. Scoping review confirming cSCC tumor cell secretion of C1r and C1s in a C1q-independent manner. Supporting source for local complement generation by cSCC tumor cells alongside Riihilä BJD 2020. Broader context for non-canonical complement pathways in cancer. Used in Section 2.4. https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1519465/full
Bennion A, Lysaght J, Lynam-Lennon N. The insider’s perspective: The intracellular complosome and immune cell dynamics in cancer. Clin Transl Med. 2026;16(2):e70628. doi: 10.1002/ctm2.70628. PMID: 41719156. PMCID: PMC12922784. February 2026 field consensus review on intracellular complement in cancer. Confirms cancer cells generate C5 and C3 intracellularly; cathepsin D cleaves intracellular C5 to generate C5a; intracellular C5aR1 named as the field’s number one therapeutic priority. C5aR1 blockade synergizes with checkpoint inhibitors. Primary source for the complosome/autocrine intracellular C5a section in 2.4. https://onlinelibrary.wiley.com/doi/10.1002/ctm2.70628
Shah HK, Prem S, Wu X, Liszewski MK, Atkinson JP, Singh AK, Kapoor V. Complement system in cancer: friend or foe of immunotherapy. J Immunother Cancer. 2026;14(3):e013290. doi: 10.1136/jitc-2024-013290. PMID: 41819546. PMCID: PMC12983960. March 2026 review confirming C5aR1 inhibition synergizes with checkpoint inhibitors. Documents tumor upregulation of complement regulatory proteins to evade surface complement attack. Used in Section 3. https://jitc.bmj.com/content/14/3/e013290
Hudgins H, Molina V, Wiernicki S, Okwuegbe K, Feng X, Wang H. Targeting C3a and C5a Signaling — A Game Changer for Cancer Therapy? Biology (Basel). 2025;14(11):1491. doi: 10.3390/biology14111491. PMID: 41300283. PMCID: PMC12649891. 2025 systematic review confirming C5a as a cancer progression driver across tumor types including cSCC and HNSCC. Used in Section 2.6. https://www.mdpi.com/2079-7737/14/11/1491
Sections 1.1, 3 & 8 — Epidemiology and Market
Jiang R, Fritz M, Que SKT. Cutaneous Squamous Cell Carcinoma: An Updated Review. Cancers (Basel). 2024;16(10):1800. doi: 10.3390/cancers16101800. PMID: 38791879. PMCID: PMC11119634. Published 8 May 2024. Open access. Source for 2.4%–5.7% annual incidence growth figure, cited from a German registry study predicting trends to 2044. Notes that US national cancer registries do not capture cSCC incidence separately. Used in Sections 1.1 and 1.2. https://pmc.ncbi.nlm.nih.gov/articles/PMC11119634/
Towards Healthcare. Global Cutaneous Squamous Cell Carcinoma Treatment Market. Published September 2025. Source for global cSCC treatment market valuation: $13.7 billion (2024), projected $27.54 billion (2034), CAGR 7.24%. Market report — commercial source, label ~. Used in Sections 1.1, 3, and 8. https://www.towardshealthcare.com/insights/cutaneous-squamous-cell-carcinoma-treatment-market-sizing
U.S. Food & Drug Administration. FDA approves cosibelimab-ipdl for metastatic or locally advanced cutaneous squamous cell carcinoma. Press release. December 13, 2024. Regulatory source for cosibelimab FDA approval. Used in Section 3. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-cosibelimab-ipdl-metastatic-or-locally-advanced-cutaneous-squamous-cell-carcinoma
Sun Pharmaceutical Industries / Checkpoint Therapeutics. Sun Pharma to Acquire Checkpoint Therapeutics. Press release. March 10, 2025. Source for Sun Pharma acquisition of Checkpoint Therapeutics and cosibelimab. Upfront consideration approximately $355 million. Used in Section 3. https://www.sec.gov/Archives/edgar/data/1651407/000110465925021976/tm258581d1_8k.htm
Sections 2.2 / 4 — Clinical Signal
Heppt M, Thielert C, Castro M, Tawfik H, Berking C. Complete Response of a Patient with Advanced Cutaneous Squamous Cell Carcinoma after Vilobelimab Monotherapy. Abstract 3369. EADV Congress 2025, Paris, 17–20 September 2025. The 79-year-old patient case. Cemiplimab failure → vilobelimab monotherapy → biopsy-confirmed complete remission at Cycle 6, maintained through Cycle 12. Overall response 512 days, at least 418 days confirmed. 46 infusions through Cycle 15. No drug-related adverse effects. Opens Section 1.1 and anchors Section 2.2. https://eadvapps.m-anage.com/eadvcongress2025/en-GB/pag/presentation/56787. Abstract (pdf) https://s3.eu-central-1.amazonaws.com/m-anage.com.storage.eadv/abstracts_congress_2025/55414.pdf
InflaRx GmbH. Non-comparative Study of IFX-1 Alone or IFX-1+Pembrolizumab in Patients With Locally Advanced or Metastatic cSCC. ClinicalTrials.gov Identifier NCT04812535. Results posted February 11, 2025. Trial record for the vilobelimab cSCC study. Monotherapy arm: 1 confirmed complete remission in 10 evaluable patients (10% rate). Source for aggregate monotherapy data cited in the competitive landscape table. https://clinicaltrials.gov/study/NCT04812535?tab=results
Sections 5 & 6 — Patient Population and Biomarker
Xiao X, Yu Q, Han B, Fu M, Chen M. Predictive role of peripheral blood indicators in the prognosis of patients with cutaneous squamous cell carcinoma treated with immune checkpoint inhibitors. Am J Cancer Res. 2025;15(4):1705–1718. PMID: 40371131. PMCID: PMC12070112. 139 cSCC patients on checkpoint inhibitors. NLR strongest single predictor of non-response (AUC 0.775). Combined model AUC 0.878 with 93% specificity. Primary source for NLR as enrollment biomarker in Sections 4 and 6. https://europepmc.org/article/MED/40371131
Section 7 — Study Design
InflaRx GmbH. InflaRx Announces Positive Topline Results from the Single Ascending Dose (SAD) Phase 1 Study with C5aR Inhibitor INF904. Press release. September 2023. Primary source for INF904 pharmacokinetic data: approximately 3-fold higher Cmax versus avacopan; approximately 10-fold higher AUClast; doubled in vivo inhibitory effect. Used in Sections 2.5 and 7. https://www.inflarx.de/Home/Investors/Press-Releases/Press-Release~2023-09-InflaRx-Announces-Positive-Topline-Results-from-the-Single-Ascending-Dose--SAD--Phase-I-Study-with-C5aR-Inhibitor-INF904~.html
Disclosure & Accuracy Note
The biological and clinical claims in this article have been cross-referenced against peer-reviewed literature, FDA communications, company disclosures, and patent filings. Interpretive claims are labeled with ~ throughout. Forward-looking claims are labeled with ?. This article represents the author’s research and analysis, not financial advice. The author may hold positions in securities discussed.